
NEXPOVIO 20 mg, comprimé pelliculé, boîte de 4 plaquettes thermoformées de 5
Dernière révision : 14/08/2023
Taux de TVA : 2.1%
Prix de vente : 5 176,28 €
Taux remboursement SS : 100%
Base remboursement SS : 5 176,28 €
Laboratoire exploitant : PHARMA BLUE
Source :

NEXPOVIO est indiqué :
• en association avec le bortézomib et la dexaméthasone pour le
traitement du myélome multiple chez les patients adultes qui ont reçu
au moins un traitement antérieur.
• en association avec la dexaméthasone pour le traitement du myélome
multiple chez les patients adultes qui ont reçu au moins quatre
traitements antérieurs et dont la maladie est réfractaire à au moins
deux inhibiteurs du protéasome, deux immunomodulateurs et un anticorps
monoclonal anti-CD38, et chez qui la maladie a progressé lors du
dernier traitement.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Pour les médicaments administrés en association avec le sélinexor, il convient de consulter le Résumé des Caractéristiques du Produit (RCP) de ces médicaments avant le début du traitement, y compris les mises en garde spéciales et précautions d'emploi, et les traitements concomitants recommandés.
Traitements concomitants recommandés
Il convient de conseiller aux patients de maintenir un apport hydrique et calorique adéquat pendant toute la durée du traitement. Pour les patients à risque de déshydratation, il convient d'envisager une hydratation par voie intraveineuse.
Il est recommandé d'instaurer un traitement prophylactique concomitant par un antagoniste 5-HT3 et/ou d'autres agents antiémétiques avant et pendant le traitement par NEXPOVIO (voir rubrique Effets indésirables).
Hématologie
La formule sanguine complète (FSC) des patients doit être contrôlée avant le traitement, pendant celui-ci et si la situation clinique l'exige. La surveillance doit être plus fréquente pendant les deux premiers mois de traitement.
Thrombopénie
Des événements liés à une thrombopénie (thrombopénie et numération plaquettaire diminuée) ont fréquemment été rapportés par les patients recevant le sélinexor ; ces événements peuvent être graves (grade 3/4). Une thrombopénie de grade 3/4 peut parfois causer des saignements cliniquement significatifs et dans de rares cas une hémorragie pouvant être fatale (voir rubrique Effets indésirables).
La thrombopénie peut être prise en charge par des interruptions du traitement, des modifications de la posologie, des transfusions de plaquettes et/ou d'autres traitements si la situation clinique l'exige. Les patients doivent bénéficier d'une surveillance des signes et des symptômes de saignement et être évalués rapidement. Pour les recommandations de modifications de la posologie, se reporter aux tableaux 1 et 2 de la rubrique Posologie et mode d'administration
Neutropénie
Une neutropénie, y compris une neutropénie grave (de grade 3/4), a été signalée chez des patients recevant le sélinexor. Dans peu de cas, des infections concomitantes ont été observées chez des patients présentant une neutropénie de grade 3/4 (voir rubrique Effets indésirables).
Les patients présentant une neutropénie doivent bénéficier d'une surveillance des signes et des symptômes d'infection et être évalués rapidement. La neutropénie peut être prise en charge par des interruptions du traitement, des modifications de la posologie et par des facteurs de croissance hématopoïétiques, conformément aux recommandations médicales. Pour les recommandations de modifications de la posologie, se reporter aux tableaux 1 et 2 de la rubrique Posologie et mode d'administration.
Toxicité gastro-intestinale
Des nausées, des vomissements et des diarrhées pouvant parfois être graves et nécessiter l'administration d'antiémétiques et d'antidiarrhéiques ont été rapportés (voir rubrique Effets indésirables).
Instaurer un traitement prophylactique par des antagonistes 5-5HT3 et/ou d'autres agents antiémétiques avant et pendant le traitement par sélinexor. Des solutés contenant des électrolytes doivent être administrés pour prévenir une déshydratation chez les patients à risque.
Les nausées/vomissements peuvent être pris en charge par une interruption du traitement ou une modification de la posologie et/ou la mise en place d'un traitement par d'autres antiémétiques, si la situation clinique l'exige. La diarrhée peut être prise en charge par une interruption du traitement ou une modification de la posologie et/ou par l'administration d'antidiarrhéiques. Pour les recommandations de modifications de la posologie, se reporter aux tableaux 1 et 2 de la rubrique Posologie et mode d'administration.
Perte de poids et anorexie
Le sélinexor peut provoquer une perte de poids et une anorexie. Le poids corporel des patients, ainsi que leur état nutritionnel et le volume de nourriture ingéré, doivent être contrôlés avant le traitement, pendant celui-ci et si la situation clinique l'exige. La surveillance doit être plus fréquente pendant les deux premiers mois de traitement. Les patients chez lesquels un appétit diminué et une perte de poids surviennent ou s'aggravent peuvent nécessiter une modification de la posologie, la prise de stimulants de l'appétit et des consultations avec un nutritionniste. Pour les recommandations de modifications de la posologie, se reporter aux tableaux 1 et 2 de la rubrique Posologie et mode d'administration.
État confusionnel et sensation vertigineuse
Le sélinexor peut provoquer un état confusionnel et une sensation vertigineuse. Il convient de demander aux patients d'éviter les situations dans lesquelles une sensation vertigineuse ou un état confusionnel peut être un problème et de ne pas prendre d'autres médicaments qui provoquent une sensation vertigineuse ou un état confusionnel sans un avis médical adéquat. Il convient de conseiller aux patients de ne pas conduire ni d'utiliser de machinerie lourde jusqu'à disparition des symptômes (voir rubrique Effets sur l'aptitude à conduire des véhicules et à utiliser des machines).
Hyponatrémie
Le sélinexor peut provoquer une hyponatrémie. Le taux de sodium des patients doit être contrôlé avant le traitement, pendant celui-ci et si la situation clinique l'exige. La surveillance doit être plus fréquente pendant les deux premiers mois de traitement. Le taux de sodium doit être corrigé en cas d'hyperglycémie concomitante (glucose sérique > 150 mg/dL) et de taux sériques de paraprotéine élevés. L'hyponatrémie doit être traitée selon les recommandations médicales (solution de chlorure de sodium par voie intraveineuse et/ou comprimés de sel), dont une consultation diététique. Les patients peuvent nécessiter une interruption du traitement par sélinexor et/ou une modification de la posologie. Pour les recommandations de modifications de la posologie, se reporter aux tableaux 1 et 2 de la rubrique Posologie et mode d'administration.
Cataracte
Le sélinexor peut provoquer l'apparition ou l'exacerbation de la cataracte (voir rubrique Effets indésirables). Un examen ophtalmologique peut être réalisé si la situation clinique l'exige. La cataracte doit être traitée selon les recommandations médicales, y compris par chirurgie si elle est garantie.
Syndrome de lyse tumorale
Un syndrome de lyse tumorale (SLT) a été rapporté chez des patients recevant le traitement par sélinexor. Les patients à haut risque de SLT doivent être étroitement surveillés. Le SLT doit être traité rapidement conformément aux recommandations médicales.
Femmes en âge de procréer/contraception chez les hommes et les femmes
Il convient de conseiller aux femmes en âge de procréer d'éviter toute grossesse ou de s'abstenir de tout rapport sexuel pendant le traitement par sélinexor et pendant au moins 1 semaine après la dernière dose de ce médicament.
Il convient de conseiller aux femmes en âge de procréer et aux hommes d'utiliser des mesures de contraception efficaces ou de s'abstenir de toute activité sexuelle pour éviter une grossesse de leur partenaire pendant le traitement par sélinexor et pendant au moins 1 semaine après la dernière dose de sélinexor (voir rubrique Fertilité, grossesse et allaitement).
Excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé de 20 mg, c.-à-d. qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
La sécurité d'emploi du sélinexor utilisé en association avec le bortézomib et la dexaméthasone a été évaluée chez 195 patients atteints d'un myélome multiple. Les effets indésirables les plus fréquents (≥ 30 %) ont été la thrombopénie (62 %), les nausées (50 %), la fatigue (42 %), l'anémie (37 %), l'appétit diminué (35 %), la diarrhée (33 %) et la neuropathie périphérique (33 %).
Les effets indésirables graves les plus fréquents (≥ 3 %) ont été la pneumonie (14,9 %), la cataracte (4,6 %), le sepsis (4,1 %), la diarrhée (3,6 %), les vomissements (3,6 %) et l'anémie (3,1 %).
La sécurité d'emploi du sélinexor utilisé en association avec la dexaméthasone a été évaluée chez 214 patients atteints d'un myélome multiple, dont 83 ayant une maladie réfractaire à une pentathérapie. Les effets indésirables les plus fréquents (≥ 30 %) ont été les nausées (75 %), la thrombopénie (75 %), la fatigue (66 %), l'anémie (60 %), l'appétit diminué (56 %), le poids diminué (49 %), la diarrhée (47 %), les vomissements (43 %), l'hyponatrémie (40 %), la neutropénie (36 %) et la leucopénie (30 %).
Les effets indésirables graves les plus fréquents (≥ 3 %) ont été la pneumonie (7,5 %), le sepsis (6,1 %) la thrombopénie (4,7 %), l'insuffisance rénale aiguë (3,7 %) et l'anémie (3,3 %).
Tableau récapitulatif des effets indésirables
Les effets indésirables rapportés dans les études cliniques sur le sélinexor en association avec le bortézomib et la dexaméthasone (SVd) sont résumés dans le Tableau 3.
Les effets indésirables rapportés dans les études cliniques sur le sélinexor en association avec la dexaméthasone (Sd) sont résumés dans le Tableau 4.
Ces effets sont présentés par classe de systèmes d'organes et fréquence. Les catégories de fréquence sont définies comme suit : très fréquent (≥1/10) ; fréquent (≥1/100, <1/10) ; peu fréquent (≥1/1 000, <1/100) ; rare (≥1/10 000, <1/1 000) ; très rare (<1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Dans chaque catégorie de fréquence, les effets indésirables sont présentés dans l'ordre décroissant de gravité.
Tableau 3 : Effets indésirables du médicament observés chez les patients atteints d'un myélome multiple traités par sélinexor en association avec le bortézomib et la dexaméthasone (SVd)
| Classe de systèmes d'organes/termepréférentiel | Tous les effets indésirables/fréquence | Effets indésirables de grade 3 et 4/fréquence |
| Infections et infestations | Très fréquent Pneumonie*, infection des voies aériennes supérieures, bronchite, rhinopharyngite Fréquent Sepsis*, infection des voies aériennes inférieures | Très fréquent Pneumonie* Fréquent Sepsis*, infection des voies aériennes inférieures, bronchite, infection des voiesaériennes supérieures |
| Affections hématologiques et du système lymphatique | Très fréquent Thrombopénie, anémie, neutropénie* Fréquent Leucopénie, lymphopénie | Très fréquent Thrombopénie, anémie Fréquent Neutropénie*, lymphopénie Peu fréquent Leucopénie |
| Troubles du métabolisme et de la nutrition | Très fréquent Appétit diminué Fréquent Hyponatrémie, déshydratation, hypokaliémie, hypocalcémie, hypophosphatémie, hyperkaliémie,hypomagnésémie | Fréquent Hyponatrémie, déshydratation, appétit diminué, hypokaliémie, hypocalcémie, hypophosphatémie |
| Affections psychiatriques | Très fréquent Insomnie Fréquent État confusionnel | Fréquent État confusionnel, insomnie |
| Affections du système nerveux | Très fréquent Neuropathie périphérique, sensation vertigineuse, céphalée Fréquent Syncope, amnésie*, trouble de l'équilibre, dysgueusie,agueusie | Fréquent Syncope, neuropathie périphérique Peu fréquent Céphalée, sensation vertigineuse, amnésie* |
| Affections de l'oreille et du labyrinthe | FréquentVertiges | Aucun |
| Classe de systèmes d'organes/terme préférentiel | Tous les effets indésirables/fréquence | Effets indésirables de grade 3 et 4/fréquence |
| Affections oculaires | Très fréquent Cataracte, vision trouble* | Très fréquent Cataracte Fréquent Vision trouble* |
| Affections cardiaques | Fréquent Tachycardie | Aucun |
| Affections vasculaires | Fréquent Hypotension | Fréquent Hypotension |
| Affections respiratoires, thoraciques et médiastinales | Très fréquent Toux Fréquent Dyspnée*, épistaxis | Fréquent Épistaxis Peu fréquent Dyspnée*, toux |
| Affections gastro-intestinales | Très fréquentNausée, diarrhée, vomissement, constipationFréquentDouleur abdominale,dyspepsie, bouche sèche, flatulence | Fréquent Nausée, diarrhée, vomissement |
| Affections de la peau et du tissu sous-cutané | Fréquent Alopécie, sueurs nocturnes*, prurit | Peu fréquent Sueurs nocturnes* |
| Affections musculo-squelettiques et systémiques | Fréquent Hypercréatinémie | Fréquent Hypercréatinémie |
| Affections du rein et des voies urinaires | Fréquent Insuffisance rénale aiguë | Fréquent Insuffisance rénale aiguë |
| Troubles généraux et anomalies au site d'administration | Très fréquent Fatigue, fièvre, asthénie Fréquent Détérioration générale de l'état de santé, malaise | Très fréquent Fatigue Fréquent Fièvre, asthénie, détérioration générale de l'état de santé |
| Investigations | Très fréquent Poids diminué Fréquent Aspartate aminotransféraseaugmentée, alanine aminotransférase augmentée | Fréquent Poids diminué, aspartate aminotransférase augmentée, alanine aminotransférase augmentée |
| Lésions, intoxications et complications liées aux procédures | Fréquent Chute, contusion | Fréquent Chute |
* Regroupement de plusieurs termes préférentiels MedDRA, dont :
- Pneumonie : pneumonie, infection pulmonaire, pneumonie à pneumocoque, pneumonie grippale, pneumonie à virus para-influenza, pneumonie bactérienne et pneumonie fongique
- Sepsis : sepsis, choc septique, sepsis staphylococcique et sepsis urinaire
- Neutropénie : neutropénie et neutropénie fébrile
- Amnésie : amnésie et atteinte de la mémoire
- Vision trouble : vision trouble, défauts visuels et baisse de l'acuité visuelle
- Dyspnée : dyspnée et dyspnée d'exercice
- Sueurs nocturnes : sueurs nocturnes et hyperhidrose
Tableau 4 : Effets indésirables du médicament observés chez les patients traités par sélinexor en association avec la dexaméthasone (Sd)
| Classe de systèmes d'organes/terme préférentiel | Tous les effets indésirables/fréquence | Effets indésirables de grade 3 et 4/fréquence |
| Infections et infestations | Très fréquent Pneumonie, infection des voies aériennes supérieures Fréquent Sepsis, bactériémie | Fréquent Pneumonie, sepsis, bactériémie Peu fréquent Infection des voies aériennes supérieures |
| Affections hématologiques et du système lymphatique | Très fréquent Thrombopénie, anémie, neutropénie, leucopénie, lymphopénie Fréquent Neutropénie fébrile | Très fréquent Thrombopénie, anémie, neutropénie, leucopénie, lymphopénie Fréquent Neutropénie fébrile |
| Troubles du métabolisme et de la nutrition | Très fréquent Hyponatrémie, déshydratation, appétit diminué, hyperglycémie, hypokaliémie Fréquent Hypocalcémie, hypophosphatémie, hyperkaliémie, hypomagnésémie, hyperamylasémie, hyperuricémie, hyperlipasémie Peu fréquent Syndrome de lyse tumorale | Très fréquent Hyponatrémie Fréquent Déshydratation, appétit diminué, hypokaliémie, hyperglycémie, hypocalcémie, hyperkaliémie, hyperamylasémie, hypophosphatémie, hyperuricémie, hyperlipasémie Peu fréquent Syndrome de lyse tumorale |
| Affections psychiatriques | Très fréquent État confusionnel, insomnie Fréquent Délire, hallucinations | Fréquent État confusionnel, insomnie Peu fréquent Délire, hallucinations |
| Affections du système nerveux | Très fréquent Sensation vertigineuse, dysgueusie, céphalée Fréquent Neuropathie périphérique, syncope, agueusie, trouble du goût, trouble de l'équilibre, trouble cognitif, perturbation de l'attention, agueusie, atteinte de la mémoire Peu fréquent Encéphalopathie | Fréquent Syncope, trouble cognitif Peu fréquent Neuropathie périphérique, encéphalopathie |
| Classe de systèmes d'organes/terme préférentiel | Tous les effets indésirables/fréquence | Effets indésirables de grade 3 et 4/fréquence |
| Affections oculaires | Très fréquent Vision trouble Fréquent Cataracte, défauts visuels | Fréquent Cataracte Peu fréquent Vision trouble, défauts visuels |
| Affections cardiaques | Fréquent Tachycardie | Aucun |
| Affectionsvasculaires | Fréquent Hypotension | Peu fréquent Hypotension |
| Affections respiratoires, thoraciques et médiastinales | Très fréquent Dyspnée, épistaxis, toux | Fréquent Dyspnée Peu fréquent Épistaxis |
| Affections gastro- intestinales | Très fréquent Nausée, diarrhée, vomissement, douleur abdominale, constipation Fréquent Dyspepsie, bouche sèche, gêne abdominale, flatulence | Fréquent Nausée, diarrhée, vomissement, constipation Peu fréquent Douleur abdominale |
| Affections de la peauet du tissus sous- cutané | FréquentAlopécie, sueurs nocturnes, prurit | Aucun |
| Affections musculo- squelettiques etsystémiques | FréquentContractures musculaires, hypercréatinémie | Peu fréquent Contractures musculaires, hypercréatinémie |
| Affections du rein etdes voies urinaires | FréquentInsuffisance rénale aiguë | Fréquent Insuffisance rénale aiguë |
| Troubles généraux et anomalies au site d'administration | Très fréquent Fatigue, fièvre, asthénie Fréquent Détérioration générale de l'état de santé, malaise, troubles de la démarche, frissons | Très fréquent Fatigue Fréquent Asthénie, détérioration générale de l'état de santé, douleur Peu fréquent Fièvre |
| Investigations | Très fréquent Poids diminué Fréquent Aspartate aminotransférase augmentée, alanine aminotransférase augmentée, phosphatase alcalinesanguine augmentée | Fréquent Alanine aminotransférase augmentée Peu fréquent Poids diminué, aspartate aminotransférase augmentée |
| Lésions, intoxications et complications liéesaux procédures | Fréquent Chute | Fréquent Chute |
Description de certains effets indésirables
Infections
L'infection a été la toxicité non hématologique la plus fréquente.
Chez les patients traités par SVd, des infections ont été rapportées chez 70 % d'entre eux ; chez 28 % des patients, ces infections ont été de grade 3 ou 4. Des infections graves ont été rapportées chez 28 % des patients et des infections fatales chez 4 % des patients traités. L'infection des voies aériennes supérieures et la pneumonie ont été les infections les plus fréquemment rapportées, chez 21% et 15 % des patients, respectivement. L'infection a conduit à un arrêt du traitement chez 1 % des patients, à une interruption du traitement chez 48 % des patients et à une réduction de la posologie chez 10 % d'entre eux.
Chez les patients traités par Sd, des infections ont été rapportées chez 53 % des patients. Parmi ces infections, 22 % ont été de grade 3 ou 4. L'infection des voies aériennes supérieures et la pneumonie ont été les infections les plus fréquemment rapportées (chez 15 % et 13 % des patients, respectivement), 25 % des infections rapportées ayant été graves et 3 % des patients traités ayant présenté une infection fatale. L'infection a conduit à un arrêt du traitement chez 7 % des patients, à une interruption du traitement chez 19 % des patients et à une réduction de la posologie chez 1 % d'entre eux.
Thrombopénie
Chez les patients traités par SVd, une thrombopénie est survenue chez 62 % d'entre eux et chez 41 % des patients, cette thrombopénie a été de grade 3 ou 4. La thrombopénie a été grave chez 2 % des patients. Sur les 41 % de patients dont la thrombopénie était de grade 3 ou 4, des saignements concomitants de grade 3 ou supérieur (concomitance définie comme la survenue dans les ± 5 jours) ont été rapportés chez 5 % des patients. Une hémorragie fatale s'est produite chez 2 % des patients atteints de thrombopénie. La thrombopénie a conduit à un arrêt du traitement chez 2 % des patients, à une interruption du traitement chez 35 % des patients et à une réduction de la posologie chez 33 % d'entre eux.
Chez les patients traités par Sd, une thrombopénie est survenue chez 75 % des patients et 65 % de ces effets indésirables du médicament étaient de grade 3 ou 4. La thrombopénie a été grave chez 5 % des patients. Sur les 65 % de patients dont la thrombopénie était de grade 3 ou 4, des saignements concomitants graves/de grade 3 ou supérieur (concomitance définie comme la survenue dans les ± 5 jours) ont été rapportés chez 5 % des patients. La thrombopénie a conduit à un arrêt du traitement chez 3 % des patients, à une interruption du traitement chez 22 % des patients et à une réduction de la posologie chez 32 % d'entre eux.
La thrombopénie peut être prise en charge par des modifications de la posologie (voir rubrique Posologie et mode d'administration), des soins de soutien et des transfusions de plaquettes. Les patients doivent bénéficier d'une surveillance des signes et des symptômes de saignement et être évalués rapidement (voir rubrique Mises en garde spéciales et précautions d'emploi).
Neutropénie
Chez les patients traités par SVd, une neutropénie est survenue chez 16 % d'entre eux et chez 10 % des patients, les événements de neutropénie ont été de grade 3 ou 4. La neutropénie a été grave chez 1 % des patients. Aucun des patients n'a nécessité un arrêt du traitement dû à la neutropénie, mais celle-ci a conduit à une interruption du traitement chez 9 % des patients et à une réduction de la posologie chez 5 % d'entre eux.
Une neutropénie fébrile, rapportée comme grave, est survenue chez un patient (< 1 %) traité par SVd ; elle était de grade 4. Elle a conduit à une interruption du traitement et à une réduction de la posologie ; l'arrêt du traitement n'a pas été nécessaire. Sur les 19 patients dont la neutropénie était de grade 3 ou supérieur, des infections concomitantes graves de grade 3 ou supérieur (concomitance définie comme la survenue dans les ±5 jours) ont été rapportées chez 3 patients (16 %). Les infections concomitantes de grade 3 ou supérieur ont été les suivantes : infection des voies aériennes inférieures, bronchite et infection de l'oreille (1 patient pour chaque infection).
Une neutropénie fébrile est survenue chez 3 % des patients ; tous les cas ont été de grade 3 ou 4. Une neutropénie fébrile grave a été rapportée chez 2 % des patients, qui a conduit à un arrêt du traitement, à une interruption du traitement ou à une réduction de la posologie chez moins de 1 % des patients (pour chaque situation). Sur les 53 patients dont la neutropénie était de grade 3 ou supérieur, des infections concomitantes graves/de grade 3 ou supérieur (concomitance définie comme la survenue dans les ± 5 jours) ont été rapportées chez 6 patients (11 %). Les infections concomitantes de grade 3 ou supérieur les plus fréquemment rapportées ont été une infection des voies urinaires (3 patients) et un sepsis (2 patients).
Anémie
Chez les patients traités par SVd, une anémie est survenue chez 37 % d'entre eux et 16 % des cas étaient de grade 3. Aucun patient n'a rapporté d'anémie de grade 4 ou 5. L'anémie a été grave chez 3 % des patients. L'anémie a conduit à un arrêt du traitement chez 1 % des patients, à une interruption du traitement chez 6 % des patients et à une réduction de la posologie chez 3 % d'entre eux.
Chez les patients traités par Sd, une anémie est survenue chez 61 % des patients et 44 % des cas étaient de grade 3 ou 4. L'anémie a été grave chez 3 % des patients. L'anémie a conduit à un arrêt du traitement chez moins de 1 % des patients, à une interruption du traitement chez 4 % des patients et à une réduction de la posologie chez 1 % d'entre eux.
L'anémie peut être prise en charge par des modifications de la posologie (voir rubrique Posologie et mode d'administration) et par des transfusions sanguines et/ou l'administration d'érythropoïétine, conformément aux recommandations médicales. Pour les recommandations de modifications de la posologie, se reporter au Tableau 2 de la rubrique Posologie et mode d'administration.
Toxicité gastro-intestinale
Chez les patients traités par SVd, des nausées sont survenues chez 50 % d'entre eux et chez 8 % des patients, ces nausées ont été de grade 3 ou 4. Les nausées ont été graves chez 2 % des patients.
Lorsqu'un traitement antiémétique a été administré, le délai médian d'amélioration des nausées a été de 10 jours. Les nausées ont conduit à un arrêt du traitement chez 3 % des patients, à une interruption du traitement chez 7 % des patients et à une réduction de la posologie chez 7 % d'entre eux.
Des vomissements ont été rapportés chez 21 % des patients traités par SVd et ces vomissements ont été de grade 3 chez 4 % des patients. Aucun patient n'a rapporté de vomissements de grade 4. Les vomissements ont été graves chez 4 % des patients. Les vomissements ont conduit à un arrêt du traitement chez 2 % des patients, à une interruption du traitement chez 3 % des patients et à une réduction de la posologie chez 3 % d'entre eux.
Une diarrhée est survenue chez 33 % des patients traités par SVd et chez 7 % des patients, cette diarrhée a été de grade 3 ou 4. La diarrhée a été grave chez 4 % des patients. Elle a conduit à un arrêt du traitement chez 1 % des patients, à une interruption du traitement chez 8 % des patients et à une réduction de la posologie chez 2 % d'entre eux.
Chez les patients traités par Sd, des nausées/vomissements sont survenus chez 79 % des patients et 10 % des cas étaient de grade 3 ou 4 ; 3 % des patients ont présenté des formes graves. Lorsqu'un traitement antiémétique a été administré, le délai médian d'amélioration des nausées ou des vomissements a été de 3 jours. Les nausées/vomissements ont conduit à un arrêt du traitement chez 5 % des patients, à une interruption du traitement chez 8 % des patients et à une réduction de la posologie chez 5 % d'entre eux.
Hyponatrémie
Chez les patients traités par SVd, une hyponatrémie est survenue chez 8 % des patients et chez 5 % d'entre eux, cette hyponatrémie a été de grade 3 ou 4. L'hyponatrémie a été grave chez < 1 % des patients. La plupart des cas d'hyponatrémie étaient asymptomatiques. Aucun cas de convulsions concomitantes n'a été rapporté. L'hyponatrémie n'a pas conduit à un arrêt du traitement, mais à une interruption chez < 1 % des patients et à une réduction de la posologie chez 1 % d'entre eux.
Chez les patients traités par Sd, une hyponatrémie est survenue chez 40 % des patients et 24 % des cas étaient de grade 3 ou 4. L'hyponatrémie a été grave chez 3 % des patients. La plupart des cas d'hyponatrémie étaient asymptomatiques. Aucun cas de convulsions concomitantes n'a été rapporté.
L'hyponatrémie n'a pas conduit à un arrêt du traitement, mais à une interruption chez 6 % des patients et à une réduction de la posologie chez 1 % d'entre eux.
Cataracte
Chez les patients traités par SVd, l'incidence de l'apparition ou de l'aggravation de cataractes nécessitant une intervention clinique a été rapportée chez 24 % des patients. Le délai médian jusqu'à la nouvelle apparition de cataracte a été de 233 jours. Le délai médian jusqu'à l'aggravation de la cataracte chez les patients déjà atteints d'une cataracte à l'instauration du traitement par sélinexor a été de 261 jours (SVd). La cataracte n'a pas conduit à un arrêt du traitement, mais à une interruption chez 4 % des patients et à une réduction de la posologie chez 3 % d'entre eux. La cataracte doit être traitée selon les recommandations médicales, y compris par chirurgie si elle est garantie (voir rubriques Mises en garde spéciales et précautions d'emploi et Posologie et mode d'administration).
Syndrome de lyse tumorale
Un syndrome de lyse tumorale (SLT) est survenu chez un patient traité par Sd (< 1 %). Il a été considéré comme étant de grade 3 et grave. Les patients à haut risque de SLT doivent être étroitement surveillés. Le SLT doit être traité rapidement conformément aux recommandations médicales (voir rubrique Mises en garde spéciales et précautions d'emploi).
Personnes âgées
Parmi les patients atteints d'un myélome multiple traités par SVd, 56 % étaient âgés de 65 ans et plus et 17 % étaient âgés de 75 ans et plus. En comparant les patients âgés de 65 ans et plus aux patients plus jeunes, ce groupe de patients plus âgés affichait une incidence d'interruption du traitement en raison d'un effet indésirable plus élevée (28 % contre 13 %) et d'effets indésirables graves plus élevée (57 % contre 51 %).
Parmi les patients atteints d'un myélome multiple traités par Sd, 47 % étaient âgés de 65 ans et plus et 11 % étaient âgés de 75 ans et plus. En comparant les patients âgés de 75 ans et plus aux patients plus jeunes, ce groupe de patients plus âgés affichait une incidence d'interruption du traitement en raison d'un effet indésirable plus élevée (52 % contre 25 %), une incidence d'interruption du traitement en raison d'effets indésirables graves plus élevée (74 % contre 59 %) et une incidence d'interruption du traitement en raison d'effets indésirables mortels également plus élevée (22 % contre 8 %).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT l'initiation du traitement,
vérifier l'absence de grossesse chez les femmes en âge de procréer par un
test.
- Informer les
patients de maintenir un apport hydrique et calorique adéquat pendant toute la
durée du traitement.
- Instaurer un traitement prophylactique concomitant par un
antagoniste 5-HT3 et/ou d'autres agents antiémétiques avant et pendant le
traitement.
SURVEILLANCE AVANT ET PENDANT LE TRAITEMENT :
- Poids corporel des patients, ainsi que leur état nutritionnel
et le volume de nourriture ingéré, avant le traitement, pendant celui-ci (plus
fréquente pendant les deux premiers mois) et si la situation clinique
l'exige.
- Formule sanguine complète (FSC) avant
le traitement, pendant celui-ci (plus fréquente pendant les deux premiers mois)
et si la situation clinique l'exige.
- Taux de sodium avant
le traitement, pendant celui-ci
(plus fréquente pendant les deux premiers mois) et si la situation clinique
l'exige.
- Signes et symptômes de saignement.
Maintenir un apport hydrique et calorique adéquat pendant toute la durée du traitement.
INFORMER IMMEDIATEMENT un médecin
en cas de :
- Bleus facilement présents ou plus nombreux que d'habitude.
-
Modifications de la peau qui apparaissent comme une éruption cutanée sous forme
de petits points violets ou rougeâtres.
- Saignements prolongés après une
coupure.
- Saignements de nez ou des gencives.
- Sang dans les urines ou
les selles.
- Fièvre.
- Fatigue récente ou qui s'aggrave.
- Nausées,
vomissements, diarrhée, altération du goût, douleur abdominale,
constipation.
- Appétit et/ou poids diminué.
- Etat confusionnel et
sensation vertigineuse.
- Modifications de la vision: vision double,
sensibilité à la lumière ou éblouissement.
- Pneumonie, infection des voies
respiratoires supérieures, bronchite, difficulté respiratoire, toux.
-
Infection virale du nez et de la gorge.
- Picotements et un engourdissement
des pieds et des mains.
- Maux de tête.
- Déshydratation.
- Perte du sommeil.
- Perte d'énergie.
Patientes
et patients en âge de procréer :
UTILISER une méthode de contraception efficace ou s'abstenir de tout rapport
sexuel au cours du traitement et pendant au moins 1 semaine après la dernière
dose du médicament, pour éviter une
grossesse.
Patientes allaitantes : interrompre l'allaitement au cours
du traitement et pendant 1 semaine après la prise de la dernière dose du
médicament.
NE PAS CONDUIRE de véhicules NI UTILISER de machines en cas de :
fatigue, état confusionnel et sensation vertigineuse.
Femmes en âge de procréer/contraception chez les hommes et les femmes
Il convient de conseiller aux femmes en âge de procréer d'éviter toute grossesse ou de s'abstenir de tout rapport sexuel pendant le traitement par sélinexor et pendant au moins 1 semaine après la dernière dose de ce médicament. Un test de grossesse est recommandé pour les femmes en âge de procréer avant l'instauration du traitement par sélinexor.
Il convient de conseiller aux femmes en âge de procréer et aux patients de sexe masculin en âge de procréer d'utiliser des mesures de contraception efficaces ou de s'abstenir de toute activité sexuelle pour éviter une grossesse de leur partenaire pendant le traitement par sélinexor et pendant au moins 1 semaine après la dernière dose de ce médicament.
Grossesse
Il n'existe pas de données sur l'utilisation de sélinexor chez la femme enceinte. Les études effectuées chez l'animal ont mis en évidence que le sélinexor peut être nocif pour le foetus (voir rubrique Données de sécurité préclinique). Le sélinexor n'est pas recommandé pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception.
Si la patiente tombe enceinte pendant la prise de sélinexor, le sélinexor doit être immédiatement arrêté et la patiente doit être informée du risque potentiel pour le foetus.
Allaitement
On ne sait pas si le sélinexor ou ses métabolites sont excrétés dans le lait maternel. Un risque pour les enfants allaités ne peut être exclu. L'allaitement doit être interrompu au cours du traitement avec sélinexor et pendant 1 semaine après la prise de la dernière dose de ce médicament.
Fertilité
Selon les résultats des études animales, le sélinexor peut altérer la fertilité chez l'homme et chez la femme (voir rubrique Données de sécurité préclinique).
Aucune étude clinique d'interaction médicamenteuse dédiée n'a été réalisée.
L'utilisation concomitante d'un puissant inducteur du CYP3A4 peut conduire à une plus faible exposition au sélinexor.
Aucune différence cliniquement significative dans la pharmacocinétique du sélinexor n'a été observée en cas d'administration concomitante avec un puissant inhibiteur du CYP3A4, la clarithromycine (500 mg par voie orale, deux fois par jour, pendant 7 jours).
Aucune différence cliniquement significative sur la pharmacocinétique du sélinexor n'a été observée en cas d'administration concomitante avec des doses de paracétamol allant jusqu'à 1 000 mg par jour.
Le traitement doit être initié et supervisé par des médecins expérimentés dans la prise en charge du myélome multiple.
Posologie
Sélinexor en association avec le bortézomib et la dexaméthasone (SVd)
Les doses recommandées de sélinexor, bortézomib et dexaméthasone sur la base d'un cycle de 35 jours sont les suivantes :
· Sélinexor 100 mg par voie orale, une fois par semaine, le jour 1 de chaque semaine. La dose de sélinexor ne doit pas dépasser 70 mg/m2 par dose.
· Bortézomib 1,3 mg/m2, par voie sous-cutanée, une fois par semaine le jour 1 de chaque semaine, pendant 4 semaines, suivie d'une semaine d'arrêt.
· Dexaméthasone 20 mg par voie orale, deux fois par semaine, les jours 1 et 2 de chaque semaine.
Le traitement par sélinexor, associé au bortézomib et à la dexaméthasone doit être poursuivi jusqu'à la progression de la maladie ou jusqu'à l'apparition d'une toxicité inacceptable.
Sélinexor en association avec la dexaméthasone (Sd)
Les doses initiales recommandées de sélinexor et de dexaméthasone sont les suivantes :
· 80 mg de sélinexor par voie orale les jours 1 et 3 de chaque semaine.
· 20 mg de dexaméthasone par voie orale les jours 1 et 3 de chaque semaine, pris avec le sélinexor.
Le traitement par sélinexor associé à la dexaméthasone doit être poursuivi jusqu'à la progression de la maladie ou jusqu'à l'apparition d'une toxicité inacceptable.
Pour des informations supplémentaires sur la posologie de médicaments administrés avec NEXPOVIO, se reporter au Résumé des Caractéristiques du Produit (RCP) de ces médicaments.
Retard ou omission de prise
Si une dose de sélinexor a été oubliée ou sa prise retardée, ou si un patient vomit après avoir pris une dose de sélinexor, il ne doit pas prendre à nouveau cette dose. Les patients doivent prendre la dose suivante le jour prévu.
Modifications posologiques
Les modifications posologiques recommandées de NEXPOVIO en cas de survenue d'effets indésirables sont présentées dans le Tableau 1 et le Tableau 2.
Pour des informations supplémentaires sur la modification de la posologie des médicaments administrés avec NEXPOVIO, se reporter au Résumé des Caractéristiques du Produit (RCP) correspondant.
Tableau 1 : Étapes prédéfinies de modifications posologiques en cas d'effets indésirables
| | Sélinexor en association avec le bortézomib et ladexaméthasone (SVd) | Sélinexor en association avec la dexaméthasone (Sd) |
| Dose initiale recommandée | 100 mg une fois par semaine | 80 mg les jours 1 et 3 de chaque semaine (160 mg au total par semaine) |
| Première réduction | 80 mg une fois par semaine | 100 mg une fois par semaine |
| Deuxième réduction | 60 mg une fois par semaine | 80 mg une fois par semaine |
| Troisième réduction | 40 mg une fois par semaine | 60 mg une fois par semaine |
| Arrêt* | ||
* Si les symptômes ne disparaissent pas, le traitement doit être arrêté
Tableau 2 : Recommandations de modifications posologiques en cas d'effets indésirables
| Effet indésirablea | Survenue | Action |
| Effets indésirables hématologiques | ||
| Thrombopénie | ||
| Numération plaquettaire de 25 000 à moins de75 000/µL | Toutes | · Réduire le sélinexor d'un niveau de dose (se reporter au Tableau 1). |
| Numération plaquettaire de 25 000 à moins de75 000/µL avecsaignement concomitant | Toutes | · Interrompre le sélinexor.· Redémarrer le sélinexor au niveau de dose inférieur (se reporter au Tableau 1) une foisle saignement jugulé. |
| Numération plaquettaire inférieure à 25 000/µL | Toutes | · Interrompre le sélinexor.· Surveiller jusqu'à ce que la numération plaquettaire soit à nouveau au moins égale à 50 000/µL.· Redémarrer le sélinexor au niveau de dose inférieur (se reporter au Tableau 1). |
| Effet indésirablea | Survenue | Action |
| Neutropénie | ||
| Numération absolue des neutrophilesde 0,5 à 1,0 x 109/L sans fièvre | Toutes | · Réduire le sélinexor d'un niveau de dose (se reporter au Tableau 1). |
| Numération absolue des neutrophiles inférieure à 0,5 x 109/LOUNeutropénie fébrile | Toutes | · Interrompre le sélinexor.· Surveiller jusqu'à ce que la numération des neutrophiles soit à nouveau supérieure ou égale à 1,0 x 109/L.· Redémarrer le sélinexor au niveau de dose inférieur (se reporter au Tableau 1). |
| Anémie | ||
| Hémoglobine inférieure à 8,0 g/dL | Toutes | · Réduire le sélinexor d'un niveau de dose (se reporter au Tableau 1).· Administrer des transfusions sanguines et/ou d'autres traitements conformément aux recommandations cliniques. |
| Conséquences menaçant le pronostic vital (intervention urgente indiquée) | Toutes | · Interrompre le sélinexor.· Surveiller le taux d'hémoglobine jusqu'à ce qu'il soit à nouveau égal ou supérieur à8 g/dL.· Redémarrer le sélinexor au niveau de dose inférieur (se reporter au Tableau 1).· Administrer des transfusions sanguines et/ou d'autres traitements conformément aux recommandations cliniques. |
| Effets indésirables non hématologiques | ||
| Hyponatrémie | ||
| Taux de sodium inférieur ou égal à 130 mmol/L | Toutes | · Interrompre le sélinexor et prodiguer des soins de soutien appropriés.· Surveiller le taux de sodium jusqu'à ce qu'il soit à nouveau égal ou supérieur à130 mmol/L.· Redémarrer le sélinexor au niveau de dose inférieur (se reporter au Tableau 1). |
| Fatigue | ||
| Grade 2 d'une durée supérieure à 7 jours OUGrade 3 | Toutes | · Interrompre le sélinexor.· Surveiller jusqu'à ce que la fatigue revienne à un grade 1 ou au niveau de référence.· Redémarrer le sélinexor au niveau de dose inférieur (se reporter au Tableau 1). |
| Nausées et vomissements | ||
| Nausées de grade 1 ou 2 (apport alimentaire oral diminué sans perte de poids significative ni déshydratation ou malnutrition)OUVomissements degrade 1 ou 2 (5 épisodes maximum par jour) | Toutes | · Maintenir le sélinexor et débuter un traitement médicamenteux antiémétique supplémentaire. |
| Effet indésirablea | Survenue | Action |
| Nausées de grade 3 (apport alimentaire liquide ou calorique oral inadéquat)OUVomissements de grade 3 ou supérieur (6 épisodes ou plus par jour) | Toutes | · Interrompre le sélinexor.· Surveiller jusqu'à ce que les nausées ou les vomissements soient revenus à un grade 2 ou inférieur ou au niveau de référence.· Débuter un traitement médicamenteux antiémétique supplémentaire.· Redémarrer le sélinexor au niveau de dose inférieur (se reporter au Tableau 1). |
| Diarrhée | ||
| Grade 2 (augmentation de 4 à 6 selles par jour par rapport au niveau de référence) | 1ère | · Maintenir le sélinexor et débuter des soins de soutien. |
| 2ème et suivantes | · Réduire le sélinexor d'un niveau de dose (se reporter au Tableau 1).· Débuter des soins de soutien. | |
| Grade 3 ou supérieur (augmentation de 7 selles ou plus par jour par rapport au niveau de référence ; hospitalisation indiquée) | Toutes | · Interrompre le sélinexor et débuter des soins de soutien.· Surveiller jusqu'à ce que la diarrhée revienne à un grade 2 ou inférieur.· Redémarrer le sélinexor au niveau de dose inférieur (se reporter au Tableau 1). |
| Perte de poids et anorexie | ||
| Perte de poids de 10 % à moins de 20 %OUAnorexie associée à une perte de poids ou à une malnutrition significative | Toutes | · Interrompre le sélinexor et débuter des soins de soutien.· Surveiller jusqu'à ce que le poids revienne à plus de 90 % du poids de référence.· Redémarrer le sélinexor au niveau de dose inférieur (se reporter au Tableau 1). |
| Effets indésirables oculaires | ||
| Grade 2, hormis la cataracte | Toutes | · Réaliser un examen ophtalmologique.· Interrompre le sélinexor et prodiguer des soins de soutien.· Surveiller jusqu'au retour des symptômes oculaires au grade 1 ou au niveau de référence.· Redémarrer le sélinexor au niveau de dose inférieur (se reporter au Tableau 1). |
| Grade ≥ 3, hormis la cataracte | Toutes | · Arrêter définitivement le sélinexor.· Réaliser un examen ophtalmologique. |
| Autres effets indésirables non hématologiques | ||
| Grade 3 ou 4 (menaçant le pronostic vital) | Toutes | · Interrompre le sélinexor.· Surveiller jusqu'au retour à un grade 2 ou inférieur.· Redémarrer le sélinexor au niveau de dose inférieur (se reporter au Tableau 1). |
a. aNational Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03.
Populations particulières
Personnes âgées
Aucun ajustement posologique du sélinexor n'est nécessaire chez les patients de plus de 65 ans (voir rubriques Effets indésirables, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Aucun ajustement posologique du sélinexor n'est nécessaire chez les patients dont la fonction rénale est atteinte de manière légère, modérée ou sévère (voir rubrique Propriétés pharmacocinétiques). Il n'existe pas de données sur les patients atteints de maladie rénale en phase terminale ou sous hémodialyse permettant de définir une recommandation posologique.
Atteinte de la fonction hépatique
Aucun ajustement posologique du sélinexor n'est nécessaire chez les patients atteints de déficience hépatique (voir rubrique Propriétés pharmacocinétiques). Les données disponibles sur les patients atteints de déficience hépatique modérée ou sévère sont insuffisantes pour définir une recommandation posologique.
Population pédiatrique
La sécurité et l'efficacité de NEXPOVIO chez les enfants âgés de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible (voir rubriques Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Il n'existe pas d'utilisation justifiée de NEXPOVIO chez les enfants âgés de moins de 18 ans dans le traitement du myélome multiple.
Mode d'administration
NEXPOVIO est destiné à une administration par voie orale.
NEXPOVIO en association avec du bortézomib et de la dexaméthasone (SVd) doit être pris par voie orale à peu près à la même heure une fois par semaine, le jour 1 de chaque semaine.
NEXPOVIO, en association avec la dexaméthasoe (Sd), doit être pris à peu près à la même heure les jours 1 et 3 de chaque semaine.
Le comprimé doit être avalé entier avec de l'eau. Il ne doit pas être écrasé, mâché, cassé ou divisé afin d'éviter tout risque d'irritation cutanée par la substance active. Il peut être pris avec ou sans aliments.
Durée de conservation :
5 ans.
Précautions particulières de conservation :Le médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
En règle générale, les cas de surdosage ont été associés à des effets indésirables similaires à ceux rapportés aux doses standard et ont généralement été réversibles en 1 semaine.
Symptômes
Les symptômes aigus potentiels incluent nausées, vomissements, diarrhée, déshydratation et confusion mentale. Les signes potentiels incluent des taux de sodium bas, des enzymes hépatiques élevés et des numérations globulaires basses. Les patients doivent être étroitement surveillés et bénéficier de soins de soutien si nécessaire. Aucun décès dû à un surdosage n'a été rapporté à ce jour.
Traitement
En cas de surdosage, le patient doit être surveillé pour déceler tout signe ou symptôme d'effets indésirables et un traitement symptomatique approprié doit être immédiatement instauré.
Classe pharmacothérapeutique : agents antinéoplasiques, autres agents antinéoplasiques, Code ATC : L01XX66
Mécanisme d'action
Le sélinexor est un inhibiteur covalent réversible sélectif de l'export nucléaire (SINE) qui bloque spécifiquement l'exportine 1 (XPO1). La XPO1 est le principal médiateur de l'export nucléaire de nombreuses protéines cargo telles que les protéines de suppression tumorale (TSP), les régulateurs de croissance et les ARNm des protéines (oncogènes) de promotion de la croissance tumorale.
L'inhibition de la protéine XPO1 par le sélinexor conduit à une accumulation marquée des TSP dans le noyau, à l'arrêt du cycle cellulaire, à des réductions de plusieurs oncoprotéines telles que l'oncogène c-Myc et la cycline D1 et à l'apoptose des cellules cancéreuses. L'association du sélinexor et de la dexaméthasone et/ou du bortézomib a démontré des effets cytotoxiques synergiques sur le myélome multiple in vitro et une augmentation de son activité antitumorale in vivo sur des modèles xenogreffés de murins atteints de myélome multiple, y compris dans les modèles résistants aux inhibiteurs du protéasome.
Électrophysiologie cardiaque
L'effet de multiples doses de sélinexor allant jusqu'à 175 mg deux fois par semaine sur l'intervalle QTc a été évalué chez des patients atteints de cancers hématologiques lourdement prétraités. Le sélinexor n'a pas eu d'effet important (pas supérieur à 20 ms) sur l'intervalle QTc au niveau de dose thérapeutique.
Efficacité et sécurité cliniques
Sélinexor en association avec le bortézomib et la dexaméthasone (SVd) pour le traitement de patients atteints d'un myélome multiple
L'efficacité et la sécurité du sélinexor en association avec le bortézomib et la dexaméthasone ont été évaluées dans l'étude KCP-330-023 (BOSTON), une étude de phase 3, mondiale, randomisée, en ouvert, contrôlée par témoin actif, chez des patients atteints d'un myélome multiple qui ont reçu au moins un traitement antérieur. Pour l'étude BOSTON, les patients devaient être atteints d'un myélome mesurable selon les critères de l'International Myeloma Working Group (IMWG) et des preuves de la progression de la maladie pendant ou après leur traitement le plus récent devaient être documentées. Ils devaient aussi avoir reçu précédemment un traitement par un à trois protocoles thérapeutiques différents contre le myélome multiple. Les patients qui avaient reçu précédemment des inhibiteurs du protéasome (seuls ou dans un traitement en association) devaient avoir répondu au moins partiellement au traitement et attendre une période d'au moins 6 mois depuis leur dernier traitement par des inhibiteurs du protéasome. Aucun antécédent d'arrêt du traitement par le bortézomib à cause d'une toxicité de grade 3 ou supérieur n'était autorisé. Les patients devaient avoir un score d'état général de l'ECOG ≤ 2, et une fonction hépatique, rénale et hématopoïétique adéquate. Les patients atteints des pathologies suivantes ont été exclus de l'étude : amylose à chaîne légère systémique, myélome actif du système nerveux central, neuropathie périphérique de grade 2 ou supérieur, neuropathie douloureuse de grade 2, leucémie à plasmocytes, polyneuropathie, organomégalie, endocrinopathie, gammapathie monoclonale ou syndrome de changements cutanés (POEMS).
L'étude a comparé le traitement par sélinexor 100 mg une fois par semaine (administré par voie orale le jour 1 de chaque semaine) en association avec de la dexaméthasone 20 mg deux fois par semaine (administrée par voie orale aux jours 1 et 2 de chaque semaine) et du bortézomib 1,3 mg/m2 une fois par semaine (administré par voie sous-cutanée le jour 1 des semaines 1 à 4, et pas la semaine 5) [bras SVd] au traitement par le bortézomib 1,3 mg/m2 deux fois par semaine (administré par voie sous- cutanée aux jours 1, 4, 8 et 11) en association avec de la dexaméthasone 20 mg à faible dose deux fois par semaine (administrée par voie orale aux jours 1, 2, 4, 5, 8, 9, 11 et 12) pendant un cycle standard de 21 jours pour les 8 premiers cycles, suivi du traitement par le bortézomib 1,3 mg/2 une fois par semaine (administré par voie sous-cutanée le jour 1 des semaines 1 à 4, et pas la semaine 5) et de la dexaméthasone 20 mg à faible dose deux fois par semaine (administrée par voie orale aux jours 1 et 2 de chaque semaine), pendant des cycles ≥ 9 [bras Vd].
Le traitement a été poursuivi dans les deux bras jusqu'à la progression de la maladie, jusqu'au décès ou jusqu'à l'apparition d'une toxicité inacceptable. Après confirmation de la progression de la maladie (PM), les patients du bras de contrôle (Vd) ont pu changer de bras afin de recevoir le traitement à base de sélinexor, soit toutes les semaines SVd (protocole thérapeutique BOSTON), soit toutes les semaines Sd, sélinexor 100 mg une fois par semaine (jour 1 de chaque semaine) et faible dose de dexaméthasone 20 mg deux fois par semaine (jours 1 et 2 de chaque semaine).
Au total, 402 patients ont été randomisés : 195 dans le bras SVd et 207 dans le bras Vd.
Les caractéristiques de la maladie et des patients à l'inclusion sont décrites dans le Tableau 5.
Tableau 5 : Caractéristiques démographiques et de la maladie des patients atteints d'un myélome multiple réfractaire en rechute dans l'étude BOSTON (n = 402)
| Caractéristiques | SVd (n = 195) | Vd(n = 207) |
| Médiane entre le diagnostic et la randomisation, années (plage) | 3,81 (0,4 ; 23,0) | 3,59 (0,4 ; 22,0) |
| Temps écoulé depuis la fin du dernier traitement précédent, médian (plage) | 48 semaines (1 ; 1 088) | 42 semaines (2 ; 405) |
| Nombre de protocoles thérapeutiques antérieurs, moyenne (plage) | 1,7 (1 ; 3) | 1,7 (1 ; 3) |
| Nombre de traitements antérieurs (%) | ||
| 1 | 51 % | 48 % |
| 2 | 33 % | 31 % |
| 3 | 16 % | 21 % |
| Âge, médian (plage) | 66 ans (40 ; 87) | 67 ans (38 ; 90) |
| Patients âgés de moins de 65 ans, n (%) | 86 (44) | 75 (36) |
| Patients âgés de 65 à 74 ans, n (%) | 75 (39) | 85 (41) |
| Patients âgés de 75 ans et plus, n (%) | 34 (17) | 47 (23) |
| Hommes : Femmes, n (%) | 115 (59) : 80 (41) | 115 (56) : 92 (44) |
| Type de traitement antérieur, n (%) | ||
| Greffe de cellules souches | 76 (39) | 63 (30) |
| Lénalidomide dans toute association | 77 (39) | 77 (37) |
| Pomalidomide dans toute association | 11 (6) | 7 (3) |
| Bortézomib dans toute association | 134 (69) | 145 (70) |
| Carfilzomib dans toute association | 20 (10) | 21 (10) |
| Inhibiteur du protéasome dans toute association | 148 (76) | 159 (77) |
| Daratumumab dans toute association | 11 (6) | 6 (3) |
| Système de stadification international révisé à l'inclusion, n (%) | | |
| I | 56 (29) | 52 (25) |
| II | 117 (60) | 125 (60) |
| III | 12 (6) | 16 (8) |
| Inconnu | 10 (5) | 14 (7) |
| Cytogénétique haut risquea, n (%) | 97 (50) | 95 (46) |
| Score d'état général de l'ECOG : 0 à 1, n (%) | 175 (90) | 191 (92) |
a Inclut l'une des del (17p)/p53, t (14 ; 16), t (4 ; 14), ou 1q21.
Le critère d'évaluation principal était la survie sans progression (SSP), conformément aux critères de réponse uniforme de l'IMWG pour le myélome multiple, évaluée par un comité d'évaluation indépendant (CEI).
Sur la base d'une analyse intermédiaire de la SSP préplanifiée, où la limite de la SSP a été fixée (suivi médian de 15,1 mois), l'étude BOSTON a montré une amélioration statistiquement significative de la SSP dans le bras SVd par rapport au bras Vd ; rapport de risque (RR) = 0,70 (IC à 95 % : 0,53-0,93 ; p = 0,0075), SSP médiane de 13,9 mois (IC à 95 % : 11,7, non atteint) et 9,5 mois (IC à 95 % : 8,1, 10,8) dans les bras SVd et Vd, respectivement.
Une amélioration statistiquement significative a été constatée pour le taux de réponse globale (TRG) : 76,4 % dans le bras SVd contre 62,3 % dans le bras Vd, p = 0,0012. Le taux ≥ à unetrès bonne réponse partielle (TBRP) (le taux ≥TBRP comprend la réponse complète stricte [RCs], la réponse complète [RC] et la TBRP) était de 44,6 % dans bras SVd contre 32,4 % dans le bras Vd.
Le délai médian jusqu'à la réponse a été de 1,4 mois chez les patients traités par SVd et de 1,6 mois chez les patients traités par Vd. La durée médiane de la réponse (DDR), chez les patients répondant, a été de 20,3 mois et de 12,9 mois dans les bras SVd et Vd, respectivement.
Au moment de l'analyse intermédiaire de la SSP préplanifiée, 109 événements de survie globale (SG) s'étaient produits ; il y a eu 47 et 62 décès dans les bras SVd et Vd, respectivement (RR = 0,84 [IC à 95 % : 0,57 ; 1,23]). La survie globale médiane n'a pas été atteinte pour le bras SVd eta été de 25 mois pour le bras Vd.
Les résultats d'une analyse descriptive mise à jour avec un suivi médian de 22,1 mois corroborent ceux de la première principale. Les résultats d'efficacité sont illustrés dans le Tableau 6 et la Figure 1.
Tableau 6 : Résultats de l'efficacité évaluée par un comité d'évaluation indépendant dans l'étude BOSTON (suivi médian de 22,1 mois)
| | SVd (n = 195) | Vd(n = 207) |
| Survie sans progression (SSP)a | 0,71 (0,54 ; 0,93) | |
| Rapport de risque (IC à 95 %) | ||
| SSP médiane en mois (IC à 95 %) | 13,2 (11,7 ; 23,4) | 9,5 (8,1 ; 10,8) |
| Taux de réponse globale (TRG)b, n (%) | 150 (76,9) | 131 (63,3) |
| IC à 95 % | (70,4 ; 82,6) | (56,3 ; 69,9) |
| RCs | 19 (10) | 13 (6) |
| RC | 14 (7) | 9 (4) |
| TBRP | 54 (28) | 45 (22) |
| RP | 63 (32) | 64 (31) |
| Délai jusqu'à la réponse, mois (IC à 95 %) | 1,4 (1,4 ; 1,5) | 1,6 (1,5 ; 2,1) |
| Durée médiane de la réponse, mois (ICà 95 %)c | 17,3 (12,6 ; 26,3) | 12,9 (9,3 ; 15,8) |
| Survie globale(SG, suivi médian de 28,7 mois)a | | |
| Nombre d'événements, n (%) | 68 (35) | 80 (39) |
| SG médiane, mois (IC à 95 %) | 36,7 (30,2, non atteinte) | 32,8 (27,8, non atteinte) |
| Rapport de risque (IC à 95 %) | 0,88 (0,63 ; 1,22) | |
SVd = sélinexor-bortézomib-dexaméthasone, Vd = bortézomib-dexaméthasone, CRs = réponse complète stricte, RC = réponse complète, TBRP = très bonne réponse partielle, RP = réponse partielle
* Les résultats d'efficacité correspondent à une analyse descriptive basée sur la date limite de recueil des données fixée au 15 février 2021
a Le rapport de risque est basé sur le modèle à risque proportionnel stratifié de Cox ; la valeur p est basée sur un test du log-rank stratifié.
b Comprend la RCs + RC + TBRP + RP, valeur p basée sur le test Cochran-Mantel-Haenzel.
c Comprend les patients répondants qui ont atteint une RP ou mieux.
Figure 1 : Courbe de Kaplan-Meier de la SSP dans l'étude BOSTON (suivi médian de 22,1 mois)
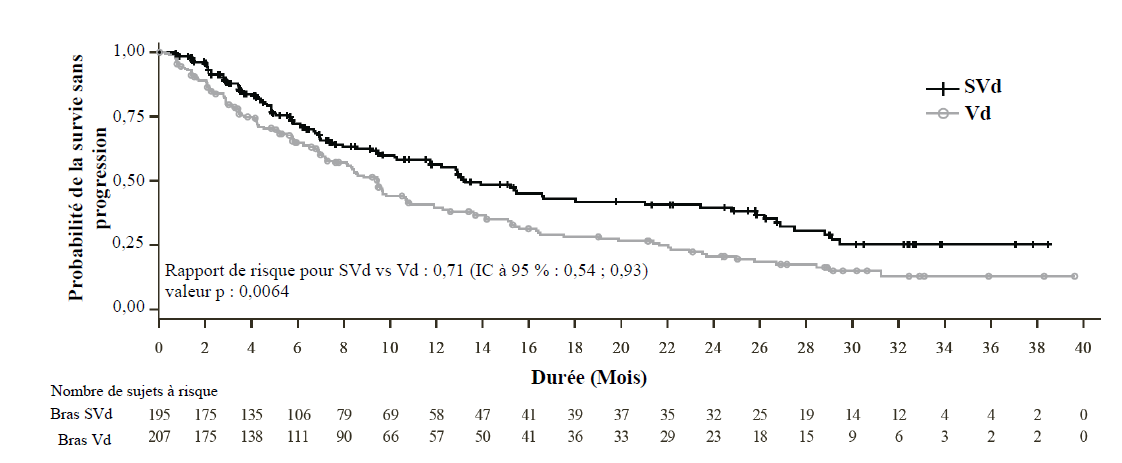
Le pourcentage de neuropathie périphérique de grade ≥ 2, un critère d'évaluation secondaire clé prédéfini, était moins élevé dans le bras SVd (21 %) que dans le bras Vd (34 %); rapport de cotes 0,50 [IC à 95 % : 0,32 ; 0,79, p = 0,0013], en raison de la dose plus faible de bortézomib administrée dans le bras SVd.
Sélinexor en association avec la dexaméthasone (Sd) pour le traitement de patients atteints d'un myélome multiple en rechute/réfractaire
L'étude KPC-330-012 (STORM), une étude de phase 2, multicentrique, à un seul bras, en ouvert, a recruté des patients atteints d'un myélome multiple réfractaire et/ou en rechute (MMRR). Pour participer à la deuxième partie de l'étude STORM, les patients devaient être atteints d'une maladie mesurable selon les critères de l'IMWG et avoir reçu précédemment au moins trois traitements contre le myélome parmi un agent alkylant, des glucocorticoïdes, du bortézomib, du carfilzomib, du lénalidomide, du pomalidomide et un anticorps monoclonal anti-CD38. Leur myélome devait être documenté comme étant réfractaire aux glucocorticoïdes, à un inhibiteur du protéasome, un immunomodulateur, un anticorps monoclonal anti-CD38 et à la dernière ligne de traitement. Les patients devaient avoir un score d'état général de l'ECOG ≤ 2, et une fonction hépatique, rénale et hématopoïétique adéquate. Les pathologies suivantes étaient synonymes d'exclusion : amylose à chaîne légère systémique, myélome actif du système nerveux central, neuropathie périphérique de grade 3 ou supérieur, neuropathie douloureuse de grade 2 ou supérieur.
Les patients ont été traités avec 80 mg de sélinexor en association avec 20 mg de dexaméthasone les jours 1 et 3 de chaque semaine. Le traitement a été poursuivi jusqu'à la progression de la maladie, jusqu'au décès ou jusqu'à l'apparition d'une toxicité inacceptable.
Parmi les patients inclus dans la partie 2 de l'étude STORM, (n = 123), quatre-vingt-trois (83) patients étaient atteints d'un MMRR réfractaire à deux inhibiteurs du protéasome (bortézomib et carfilzomib), à deux immunomodulateurs (lénalidomide et pomalodomide) et à un anticorps monoclonal anti-CD38 (daratumumab). La durée médiane du traitement par sélinexor pour ces 83 patients était de 9 semaines (plage : 1 à 61 semaines). La dose totale médiane de sélinexor reçue était de 880 mg (plage : 160 à 6 220 mg), avec une dose médiane hebdomadaire de 105 mg (plage : 22 à 180 mg).
Les données décrites ci-dessous concernent les 83 patients dont la maladie était réfractaire à la pentathérapie à base de bortézomib (B), carfilzomib (C), lénalidomide (L), pomalidomide (P) et daratumumab (D).
Le Tableau 7 présente les caractéristiques de la maladie et des traitements antérieurs des patients.
Tableau 7 : Caractéristiques démographiques et de la maladie des patients atteints d'un myélome multiple réfractaire en rechute traités par 80 mg de sélinexor et 20 mg de dexaméthasone deux fois par semaine (n = 83)
| Caractéristiques | |
| Médiane entre le diagnostic et le début du traitement de l'étude, années (plage) | 7 ans (1 ; 23) |
| Nombre de protocoles thérapeutiques antérieurs, médian (plage) | 8 (4 ; 18) |
| Âge, médian (plage) | 65 ans (40 ; 86) |
| Patients âgés de moins de 65 ans, n (%) | 40 (48) |
| Patients âgés de 65 à 74 ans, n (%) | 31 (37) |
| Patients âgés de 75 ans et plus, n (%) | 12 (15) |
| Hommes : Femmes, n (%) | 51 H (61) : 32 F (39) |
| Statut réfractaire à des associations thérapeutiques spécifiques, n (%) | |
| Réfractaire à une pentathérapie (BCLPD) | 83 (100) |
| Daratumumab dans toute association | 57 (69) |
| Daratumumab en monothérapie | 26 (31) |
| Greffe antérieure de cellules souches1, n (%)≥ 2 greffes | 67 (81)23 (28) |
| Thérapie cellulaire CAR-T antérieure, n (%) | 2 (2,4) |
| Système de stadification intégré révisé à l'inclusion, n (%) | |
| I | 10 (12) |
| II | 56 (68) |
| III | 17 (21) |
| Cytogénétique haut risque, n (%)(inclut l'une des del(17p)/p53, t(14 ; 16), t(4 ; 14), ou 1q21) | 47 (57) |
| Score d'état général de l'ECOG : 0 à 1, n (%) | 74 (89) |
1 Un patient avait eu une greffe de cellules souches allogéniques.
Le critère d'évaluation principal était le taux de réponse globale (TRG) évalué par un comité d'évaluation indépendant conformément aux critères de réponse uniforme de lIMWG pour le myélome multiple. Les réponses ont été évaluées tous les mois et selon les recommandations de l'IMWG. Le Tableau 8 donne un aperçu des résultats de l'efficacité.
Tableau 8 : Résultats de l'efficacité : évalués par un comité d'évaluation indépendant (STORM, patients atteints d'un myélome multiple réfractaire en rechute, traités par 80 mg de sélinexor et 20 mg de dexaméthasone deux fois par semaine)
| Critères d'efficacité | NEXPOVIO 80 mg+ dexaméthasone 20 mg n = 83 |
| Taux de réponse globale (TRG), n (%) (inclut RCs + TBRP + RP)1 | 21 (25,3) |
| Intervalle de confiance à 95 % | 16,4 ; 36 |
| RCs, MRM négatif, n (%) | 1 (1,2) |
| RC, n (%) | 0 (0) |
| TBRP, n (%) | 4 (4,8) |
| RP, n (%) | 16 (19,3) |
| Réponse minimale (RM), n (%) | 10 (12,0) |
| Maladie stable (MS), n (%) | 32 (38,6) |
| Progression de la maladie (PM)/Non évaluable (NE), n (%) | 20 (24,1) |
| | |
| Délai médian jusqu'à la première réponse (semaines) (plage : 1 à 10 semaines) | 3,9 |
| Durée médiane de la réponse (DDR) mois (intervalle de confiance à 95 %) | 3,8 (2,3 ; 10,8) |
1RCs = réponse complète stricte, RC = réponse complète, TBRP = très bonne réponse partielle, RP = réponse partielle
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec le sélinexor dans tous les sous-groupes de la population pédiatrique dans le traitement du MMRR (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption
Après administration orale, la concentration plasmatique maximale de sélinexor, ou Cmax, est atteinte en 4 heures. L'administration concomitante d'un repas riche en matières grasses (800 à 1 000 calories, les lipides représentant environ 50 % de la valeur énergétique totale du repas) n'a pas eu d'effet cliniquement significatif sur la pharmacocinétique du sélinexor.
Distribution
Le sélinexor est lié à 95 % aux protéines plasmatiques humaines. Dans une analyse pharmacocinétique de population, le volume apparent de distribution (Vd/F) du sélinexor était de 133 L chez les patients cancéreux.
Biotransformation
Le sélinexor est métabolisé par le CYP3A4, par plusieurs UDP-glucuronosyltransférases (UGT) et glutathione S- transférases (GST).
Élimination
Après une dose unique de 80 mg de sélinexor, la demi-vie moyenne (t½) est de 6 à 8 heures. Dans une analyse pharmacocinétique de population, la clairance apparente totale (CL/F) du sélinexor était de 18,6 L/h chez les patients cancéreux.
Populations particulières
Âge, sexe et race
L'âge (18 à 94 ans), le sexe ou la race n'ont pas eu d'effet cliniquement significatif sur la pharmacocinétique du sélinexor.
Dans le sous-ensemble pharmacocinétique de population, l'âge et la race n'étaient pas identifiés comme étant des covariables significatives. Le sexe était identifié comme étant une covariable significative.
Atteinte de la fonction rénale
Le degré d'atteinte de la fonction rénale a été déterminé par la clairance de la créatinine estimée par l'équation de Cockcroft-Gault. Les résultats des analyses pharmacocinétiques de population de patients ayant une fonction rénale normale (n = 283, CLcr : ≥ 90 mL/min), un dysfonctionnement rénal léger (n = 309, CLcr : 60 à 89 mL/min), modéré (n = 185, CLcr : 30 à 59 mL/min) ou sévère (n = 13, CLcr : 15 à 29 mL/min) indiquaient que la clairance de la créatinine n'avait pas d'impact sur les propriétés pharmacocinétiques de NEXPOVIO. Par conséquent, une atteinte de la fonction rénale légère, modérée ou sévère ne devrait pas altérer les propriétés pharmacocinétiques du sélinexor, et aucun ajustement posologique de ce médicament n'est requis chez les patients insuffisants rénaux.
Atteinte de la fonction hépatique
Une analyse pharmacocinétique de population a indiqué qu'une déficience hépatique légère (bilirubine > 1 à 1,5 x LSN ou ASAT > LSN, mais bilirubine ≤ LSN, n = 119) n'avait pas d'effet cliniquement significatif sur la pharmacocinétique du sélinexor. Des résultats similaires ont été observés chez un petit nombre de patients atteints de déficience hépatique modérée (bilirubine > 1,5 à 3 x LSN, tout taux d'ASAT, n = 10) et sévère (bilirubine > 3 x LSN, tout taux d'ASAT, n = 3).
Le sélinexor est susceptible d'avoir une influence importante sur l'aptitude à conduire des véhicules et à utiliser des machines. Le sélinexor peut provoquer de la fatigue, un état confusionnel et une sensation vertigineuse. Il convient de demander aux patients d'éviter les situations dans lesquelles une sensation vertigineuse ou un état confusionnel peut être un problème et de ne pas prendre d'autres médicaments qui provoquent une sensation vertigineuse ou un état confusionnel sans un avis médical adéquat. Il convient de conseiller aux patients de ne pas conduire ni d'utiliser des machines s'ils présentent l'un de ces symptômes.
Toxicologie en administration répétée
Les résultats de l'étude de 13 semaines d'administration d'une dose répétée chez le rat étaient des diminutions du gain pondéral et de la consommation de nourriture et une hypoplasie hématopoïétique/lymphocytaire, ainsi que des effets sur les organes de la reproduction des mâles/femelles. Dans l'étude de 13 semaines conduite chez le singe, les effets observés liés au traitement incluaient une perte de poids corporel, des effets gastro-intestinaux et une déplétion lymphocytaire/hématologique. Les toxicités gastro-intestinales, y compris l'anorexie, les diminutions du gain pondéral ainsi que la réduction de la consommation de nourriture ont été notées comme étant médiées par le SNC. Aucune marge de sécurité n'a pu être établie pour ces toxicités.
Génotoxicité
Le sélinexor n'était pas mutagène dans le test de mutation réverse sur des bactéries. Le sélinexor n'était clastogène ni dans le test cytogénétique in vitro conduit sur des lymphocytes humains, ni dans le test in vivo du micronoyau conduit chez le rat.
Carcinogénicité
Aucune étude de carcinogénicité n'a été conduite avec le sélinexor.
Toxicologie liée à la reproduction et au développement
Aucune étude de fertilité n'a été conduite chez l'animal avec le sélinexor. Dans les études de toxicité orale en administration répétée, le sélinexor a été administré pendant 13 semaines au maximum à des rats et des singes. Des diminutions du sperme, des spermatides et des cellules germinales dans les épididymes et les testicules ont été observées chez les rats, des diminutions des follicules ovariens ont également été observées chez les rates et une nécrose unicellulaire des testicules a été observée chez les singes. Ces résultats ont été observés à des expositions systémiques respectivement d'environ 0,11, 0,28 et 0,53 fois l'exposition (ASCdernière) des humains à la dose humaine recommandée de 80 mg. Des effets sur le développement ont été observés chez les rates gravides à des expositions quotidiennes systémiques inférieures à l'exposition (ASCdernière) des humains à la dose humaine recommandée de 80 mg.
Autres données de toxicologie
Un test de sensibilisation sur des cochons d'Inde a mis en évidence que le sélinexor à 25 % engendrait une réponse modérée d'hypersensibilité par une dermite de contact de grade II à 24 et 48 heures.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Médicament nécessitant une surveillance particulière pendant le traitement
Prescription hospitalière
Prescription réservée aux médecins compétents en CANCEROLOGIE
Prescription réservée aux médecins compétents en maladie du sang
Prescription réservée aux spécialistes et services HEMATOLOGIE
Prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
Remboursement en fonction de l'indication (JO du 17/09/2024) :
La seule
indication thérapeutique ouvrant droit à la prise en charge par l'assurance
maladie est :
Médicament nécessitant une surveillance particulière pendant le traitement
Prescription hospitalière
Prescription réservée aux médecins compétents en CANCEROLOGIE
Prescription réservée aux médecins compétents en maladie du sang
Prescription réservée aux spécialistes et services HEMATOLOGIE
Prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
Remboursement en fonction de l'indication (JO du 17/09/2024) :
Comprimé pelliculé
Comprimé pelliculé bleu, rond et biconvexe (4 mm d'épaisseur et de 7 mm de diamètre) portant la mention « K20 » en creux sur une face.
Comprimé pelliculé bleu, rond et biconvexe (4 mm d'épaisseur et de 7 mm de diamètre) portant la mention « K20 » en creux sur une face.
Plaquettes thermoformées en PVC/PCTFE/PVC-aluminium contenant 5 comprimés pelliculés. Un emballage extérieur contient quatre emballages intérieurs à sécurité enfant contenant chacun une plaquette. L'emballage contiennent un total de 20 comprimés pelliculés.
Chaque comprimé pelliculé contient 20 mg de sélinexor. Pour la liste complète des excipients, voir rubrique Liste des excipients.
Noyau du comprimé
Cellulose microcristalline (pH-101) (E460i)
Croscarmellose sodique (E468)
Povidone K30 (E1201)
Silice colloïdale (E551)
Stéarate de magnésium (E470b)
Cellulose microcristalline (pH-102) (E460i)
Laurilsulfate de sodium (E514i)
Enrobage du comprimé
Talc (E553b)
Poly(alcool vinylique) partiellement hydrolysé (E1203)
Monostéarate de glycérol (E471)
Polysorbate 80 (E433)
Dioxyde de titane (E171)
Macrogol (E1521)
Laque aluminique Indigotine (E132)
Laque aluminique Bleu brillant FCF (E133)